Most people assume that before a generic drug hits the shelf, it must be tested on real people. That’s not always true. The FDA allows certain generic drugs to skip human trials entirely - not because they’re cutting corners, but because science says they don’t need to.
What Exactly Is a Bioequivalence Waiver?
A bioequivalence waiver, or biowaiver, is when the FDA says: you don’t need to give this drug to volunteers to prove it works the same as the brand-name version. Instead, you can use lab tests on the pill itself. This isn’t a loophole. It’s a science-backed rule written into federal regulations (21 CFR 320.22). The goal? Save time, money, and avoid unnecessary human testing - while still making sure the generic drug does exactly what it’s supposed to.These waivers only apply to immediate-release solid oral tablets and capsules. That means no patches, no injections, no extended-release pills. If your drug dissolves quickly in the stomach and gets absorbed easily, you might qualify.
How Does the FDA Decide Who Gets a Waiver?
The answer lies in the Biopharmaceutics Classification System (BCS). It’s a simple but powerful tool that sorts drugs into four classes based on two things: how well they dissolve in water (solubility) and how easily they pass through the gut wall (permeability).Class I drugs are the golden ticket: high solubility, high permeability. Examples include metformin, atenolol, and ranitidine. For these, the FDA says: if the generic dissolves at the same rate and in the same way as the brand-name drug in a test tube, it will behave the same in the body. No blood draws needed.
Class III drugs - high solubility but low permeability - can sometimes qualify too. But the rules are tighter. The generic must use the exact same inactive ingredients (excipients) as the brand. Even a small change in filler or coating can trigger a requirement for a human study. The FDA wants to be sure the drug isn’t absorbed in a different part of the gut, which could change how it works.
Class II and IV drugs? Forget it. These are poorly soluble or poorly absorbed. For them, in vivo studies are still mandatory. That’s because their absorption depends on complex factors like stomach pH, food, or bile - things a simple dissolution test can’t predict.
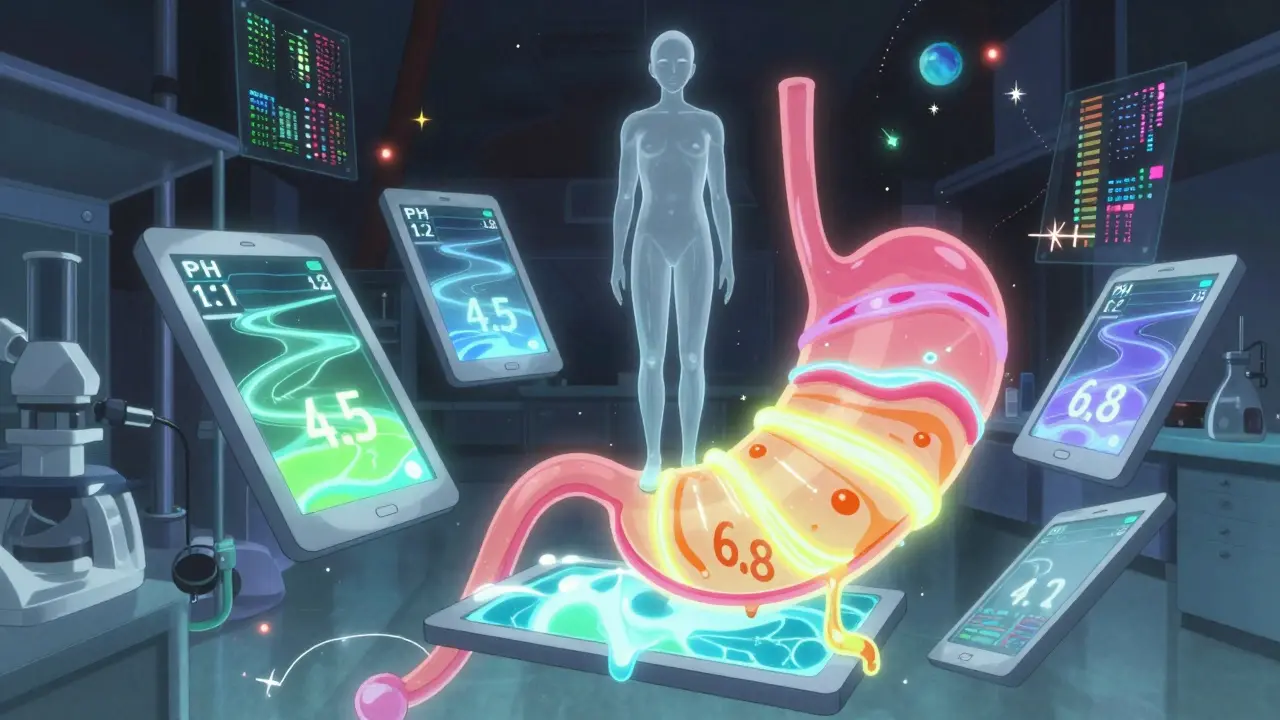
What Does the Lab Test Actually Look Like?
It’s called in vitro dissolution testing. You put 12 tablets in a machine that mimics stomach and intestinal fluids - pH 1.2 (stomach acid), pH 4.5 (early intestine), and pH 6.8 (lower intestine). The machine stirs them, and sensors track how much of the drug dissolves over time.The test must be discriminatory. That means if two pills are slightly different - say, one uses a different binder - the test should catch it. If it can’t tell them apart, the FDA rejects the waiver.
The key metric? The f2 similarity factor. It’s a number between 0 and 100. For a waiver to be approved, the generic’s dissolution profile must match the brand’s within an f2 score of 50 or higher. That’s not a guess - it’s a math-based threshold validated across hundreds of real-world cases.
Testing isn’t a quick check. It takes 60 minutes per sample, with readings at 10, 15, 20, 30, 45, and 60 minutes. That’s 12 tablets × 6 time points = 72 data points per batch. And you need to do this for at least three batches. It’s tedious, but it’s far cheaper than running a human trial.

Why Does This Matter to Patients and the System?
A typical bioequivalence study costs between $250,000 and $500,000 and takes 6 to 12 months. That’s money and time that gets passed on to patients in the form of higher drug prices or delayed access.Since 2018, biowaivers have been used in about 18% of all generic drug applications. That’s up from 12% in 2018. In 2022 alone, this shortcut saved the industry an estimated $1.2 billion in clinical trial costs, according to IQVIA. And it shaved an average of 7.3 months off approval times.
That means generics hit the market faster - and cheaper. A generic version of a common blood pressure pill might be available 8 months sooner, giving patients a $50-a-month savings instead of $300. That’s life-changing for people on fixed incomes.
Where the System Still Falls Short
The FDA’s biowaiver rules work great for simple pills. But they don’t cover most complex drugs.Modified-release tablets? No waiver. Drugs with a narrow therapeutic index - like warfarin or levothyroxine - are mostly excluded. Even though some studies show certain versions of levothyroxine could qualify, the FDA plays it safe. A tiny difference in absorption could cause serious harm.
Also, there’s inconsistency. A 2022 PhRMA survey found that 42% of companies reported different standards across FDA review divisions. One team approves a waiver for a Class III drug; another rejects it for the same formulation. That uncertainty forces companies to spend more on backup studies - adding cost and delay.
And here’s the real gap: 85% of complex generics - think patches, inhalers, or injectables - can’t use this pathway at all. The FDA admits this. Their 2023 strategic plan says they’re working to expand biowaivers, but progress is slow.

What’s Next for Bioequivalence Waivers?
The FDA is testing new approaches. A 2023 pilot program is looking at whether certain narrow therapeutic index drugs - like specific antiepileptic formulations - could qualify under stricter conditions. They’re also investing $15 million a year through GDUFA to improve dissolution methods and build better models that predict how a pill behaves in the body.Experts like Dr. Utpal Munshi, former head of FDA’s Office of Generic Drugs, call biowaivers a "scientifically rational pathway." The American Association of Pharmaceutical Scientists found that over 95% of Class I biowaiver approvals matched real-world human results.
But the real test is in the numbers. If a generic tablet dissolves just like the brand - in every buffer, at every time point - and the science says it’s absorbed the same way, why test it on people? The answer, increasingly, is: you don’t have to.
What This Means for Generic Drug Developers
If you’re developing a generic drug, the first question isn’t "How do we run a human trial?" It’s: "Is this a BCS Class I or III drug?" If yes, you start with dissolution testing - not ethics boards or recruitment ads.Successful submissions usually come from teams with at least 5 years of formulation experience. They know how to build a dissolution method that’s sensitive enough to catch differences - but not so sensitive it flags harmless variations. They also know to request a pre-ANDA meeting with the FDA. Data shows those who do have a 22% higher approval rate.
One generic company reported 12 successful biowaivers over three years. They saved $4.2 million and cut approval times by 8-10 months per drug. That’s not luck. That’s strategy.
Final Thought: Science Over Tradition
The old way of proving a drug works - testing it on humans - isn’t wrong. But it’s not always necessary. For simple, well-understood drugs, in vitro tests are more accurate, more reproducible, and more ethical.The FDA’s biowaiver system isn’t perfect. It’s limited. It’s inconsistent in places. But when it works, it’s one of the clearest examples of how science can make healthcare better - faster, cheaper, and safer.
Next time you pick up a $5 generic pill, know this: it might have never touched a human body - and that’s by design.
What drugs can get a bioequivalence waiver from the FDA?
Only immediate-release solid oral dosage forms - tablets and capsules - that meet BCS Class I (high solubility, high permeability) or sometimes Class III (high solubility, low permeability) criteria. Examples include metformin, atenolol, and ranitidine. Modified-release, injectable, or topical products do not qualify.
Do bioequivalence waivers mean the generic drug is less effective?
No. Biowaivers are granted only when in vitro data - like dissolution profiles - can reliably predict how the drug will behave in the body. For BCS Class I drugs, over 95% of approved waivers have matched real human study results. The FDA doesn’t approve these waivers lightly. They’re based on decades of research.
How much money does a bioequivalence waiver save?
A traditional bioequivalence study costs between $250,000 and $500,000 and takes 6-12 months. A biowaiver eliminates that cost entirely. Companies report saving millions across multiple products. For the industry, biowaivers have saved an estimated $1.2 billion annually in clinical trial expenses since 2020.
Can narrow therapeutic index drugs get a waiver?
Generally, no. Drugs like warfarin, levothyroxine, and cyclosporine require human studies because small differences in absorption can be dangerous. But the FDA is running a 2023 pilot program to evaluate whether certain antiepileptic drugs might qualify under strict new conditions. No broad changes have been made yet.
Why do some biowaiver applications get rejected?
The most common reason is inadequate dissolution testing. About 35% of rejected applications fail because the test method isn’t discriminatory enough - it can’t tell the difference between the generic and brand-name drug. Other reasons include using wrong pH conditions, not testing enough batches, or failing to meet the f2 similarity factor of 50 or higher.
Is the biowaiver system the same in other countries?
Yes. The FDA’s approach aligns with the International Council for Harmonisation (ICH) M9 guideline, adopted in the U.S. in January 2021. The European Medicines Agency (EMA) and Health Canada use similar BCS-based frameworks. The rules are nearly identical worldwide, making biowaivers a global standard for simple oral generics.
What’s the future of bioequivalence waivers?
The FDA aims to expand biowaivers to more drug classes by 2027, including some BCS Class III drugs and possibly narrow therapeutic index medications. They’re investing in better in vitro-in vivo correlation models and dissolution methods. Industry analysts predict biowaivers could cover 25-30% of all generic applications by 2027, up from 18% today.
Shanahan Crowell
January 2, 2026 AT 08:35Wow, this is wild-I had no idea generics could skip human trials entirely! I always assumed every pill had to be tested on people first. Turns out, science is way smarter than I thought. 😅
Kerry Howarth
January 2, 2026 AT 09:03This makes perfect sense. If a drug dissolves identically in controlled conditions, and its BCS class is well-established, human trials are redundant.
Joy F
January 3, 2026 AT 13:17Let’s be real-this isn’t science. It’s corporate cost-cutting dressed up as innovation. They’re trading patient safety for quarterly earnings. Class I drugs? Sure. But what about the 15% of people who metabolize differently? You think a dissolution test catches that? Nope. It’s a regulatory loophole with a fancy acronym. BCS? More like B.S. Culture.
Haley Parizo
January 4, 2026 AT 11:32They call it 'science'-but it’s capitalism in a lab coat. The FDA isn’t protecting patients; they’re protecting Big Pharma’s profit margins by letting generics bypass the one thing that actually proves safety: human experience. We’ve been lied to. This isn’t progress-it’s erosion. And don’t tell me '95% match' means safe. That 5%? Could be your kid. Could be your mom. Could be you.
Michael Burgess
January 6, 2026 AT 04:03As someone who’s been on metformin for 12 years-this is awesome news. 💯 I’ve been taking the same generic since 2012, and it’s never flaked. If the pill dissolves the same in the test tube, why risk a needle in someone’s arm? Also, the f2 factor? That’s the real MVP. Nerdy? Yes. Brilliant? Also yes. 🤓
Liam Tanner
January 8, 2026 AT 01:19Interesting read. I’ve always wondered how generics get approved so fast. This explains a lot. Thanks for breaking it down clearly.
Lori Jackson
January 9, 2026 AT 00:38Of course the FDA allows this. They’re bought and paid for by pharmaceutical conglomerates. The same people who pushed OxyContin now want you to believe a dissolution test is enough for your thyroid medication. Class I? Ha. What about bioaccumulation? What about gut microbiome interference? You think a pH 6.8 buffer accounts for that? Wake up. This isn’t regulation-it’s surrender.
Wren Hamley
January 9, 2026 AT 19:01So… if a pill dissolves like the brand, it’s ‘the same’? But what if the binder causes a slow-release effect in some people? Or the dye triggers a reaction? Dissolution ≠ absorption. The FDA’s model is elegant-but oversimplified. I get the savings. But I also get the fear. We’re betting lives on math. That’s… bold.
Ian Ring
January 10, 2026 AT 01:48Interesting stuff-though I must say, the f2 factor being set at 50 seems rather arbitrary. I’d like to see more peer-reviewed validation across diverse populations. Also, the EMA’s alignment is reassuring, but consistency across FDA divisions? Hmm. Not so much. 😐
JUNE OHM
January 11, 2026 AT 11:38THIS IS A GLOBALIST SCHEME TO DESTROY AMERICAN HEALTHCARE. THEY’RE LETTING CHINA MAKE OUR PILLS AND SKIPPING TESTS BECAUSE THEY’RE TOO LAZY TO DO REAL SCIENCE. THE FDA IS A CORRUPT TOOL OF THE NEW WORLD ORDER. #STOPBIOEQUIVALENCEFAKEWASERS
Philip Leth
January 11, 2026 AT 13:26Man, I just picked up my $4 generic lisinopril yesterday. Never knew it never touched a human. Kinda cool, honestly. Feels like science actually working for the people.
Angela Goree
January 13, 2026 AT 03:48So you’re telling me my blood pressure med-same chemical, same dissolution curve-was never tested on a single human? That’s not science. That’s negligence. And now I’m supposed to trust this? No. No. NO.
Tiffany Channell
January 14, 2026 AT 05:45Of course they approve waivers for Class III drugs. They don’t care about the 12% of patients who absorb it 40% slower. They care about the bottom line. And you? You’re just a data point in their spreadsheet. Wake up. This isn’t innovation-it’s exploitation.